title: “PAM Protocol” output: html_document —
Diving PAM II Basic Operation and Protocol
PAM Overview 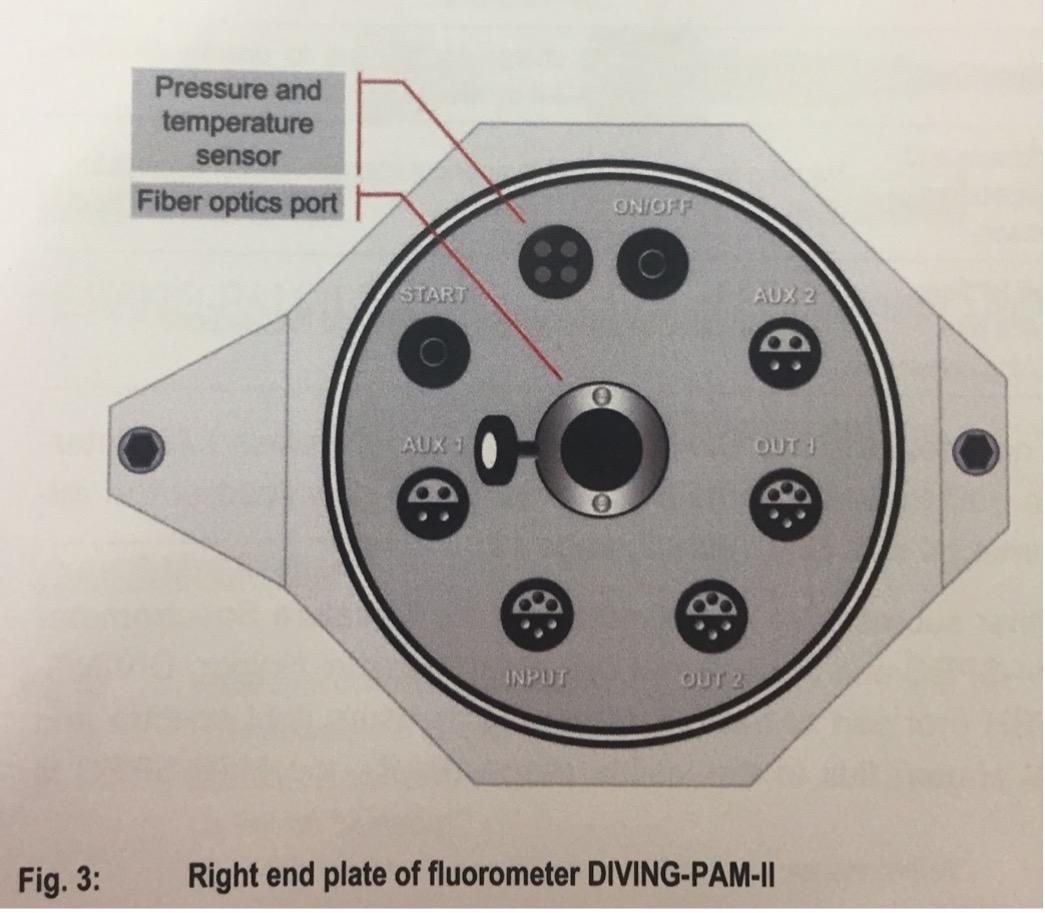
• AUX1/AUX2 connects to Mini Spec or Fiber Optic Oxygen Meter FireSting O2 (← THIS IS AMAZING)
• Fiber Optics Port: Where the fiber optics cable is inserted into the main console.
• INPUT: Interface to charge the battery and to operate the Diving PAM via WinControl-3
• As a note, this protocol as it stands will not cover operating DIVING PAM with WinControl. This protocol is meant to be optimized for in situ measurements
Connecting the Fiber Optic Cord to the PAM: Ensure it is snugly in its port and tighten screw accordingly. There should be no wiggle or play.

Setup for Leafy Material: Seagrasses, Algae, Kelp

Setup for Coral including diagrams for achieving optimal distance.
Additional Image of Coral Surface Holder with Dark Adaption

*Note the adaptor is placed directly over the surface holder. There is a slit in the black plug for the fiber optic cord to go through. The black plug attached to the string is buoyant and magnetic. It is meant for ease of use.
Getting the PAM Ready to Rumble
• Unless you are using the MINI-Spec, make sure the PAR is selected for internal PAR sensor. Select Menu ⇒ Sensors ⇒ Use Int. PAR ⇒ On Basic Data Page, select Act.L.
• Adjust GAIN: This is the first of three parameters used to optimize your signal:noise ratio. Noise increases with increasing signal amplification. *Note, Gain should be optimized to your weakest signal (i.e. your most stressed animal).
• Gain is adjusted by referencing your Ft value (i.e. your current fluorescence).
• When adjusting Gain, you want your Ft value to be within range of 300-500.
• To adjust GAIN: Select Menu ⇒ PAM Settings ⇒ Gain ⇒ Select and adjust values between 1-4.
• Adjust DAMPING: Damping suppresses High Frequency Noise. This ranges from 0-20. The higher your damping, the longer your response time.
• Adjusting DAMP vs.GAIN requires play. You will have to optimize this to the values you think are most appropriate for your organism.
• Final range of Ft should be between 300-500. Make sure you record your Gain and Damp. This is oftentimes required to publish. Also, once these values are SET, you cannot change them for the duration of your experiment and data collection period.
• Adjust F-Offset: This determines the background signal for the subtraction from the total signal.
• You MUST F-offset after every change to DAMP and GAIN. You must also adjust F-Offset before EVERY data recording period. This should be done in as close to experimental/field conditions as possible. Ideally, this should be done using experimental treatment or on DIVE in the field.
• Best practice is to ZERO PAM, using a leaf clamp with the metal slider closed.
• Select Menu ⇒ PAM Settings ⇒ Adjust F-Offset.
• It will run for 30s.
• Ft should drop to ~0. Likely between 0-20 with clip on.
make to a table
PAM SETTINGS FOR DIFFERENT CORAL SPECIES SPECIES GAIN DAMPING LIGHT SAT SPACE (mm) Agaricia spp. 2 1 10 7 Acropora cervicornis 1 1 10 7 Diploria labyrinthisformis 3 1 10 7 Siderastrea siderea 1 1 10 7 Porites porites 1 1 10 7 Montastrea cavernosa 2 1 10 7 Orbicella faveolata 1 1 8 7
Taking Measurements Fv/Fm
• Before taking measurements, the organisms should be in its natural experimental condition (light, temp, etc) for a minimum of 24 hours. This is because you want all active photosystems active. Dark Adaptation should last for 20 mins, so the photosystems are most relaxed.
• Select F2. Fv/Fm will show on the main panel.
• Cross validate your values with reported values in the literature.
• Temperature and PAR are the two most important drivers for photosynthesis. In order to make direct comparisons, conditions must be the same or as similar as possible.
Rapid Light Curves
• Again, this requires some play and a defined expectation for outcome. Literature searches are critical to determine the duration of the light curve.
• RLC should start at a PAR value below that of its natural environment. They should be acclimated to experimental conditions and NOT dark adapted.
• If the illumination steps are long enough and refined enough to reach photosynthetic steady state (the asymptote), you can use them as classical light response curves. This means you can calculate alpha, rETRMAX , Ek with considerable confidence.
References
Tunala, Layla Poubel, Frederico T.S. Tâmega, Heitor M. Duarte, and Ricardo Coutinho. 2019. Stress Factors in the Photobiology of the Reef Coral Siderastrea Stellata. Journal of Experimental Marine Biology and Ecology. 519: 151188. https://doi.org/10.1016/j.jembe.2019.151188.
Manzello, D., M. Warner, E. Stabenau, J. Hendee, M. Lesser, and M. Jankulak. 2009. Remote Monitoring of Chlorophyll Fluorescence in Two Reef Corals during the 2005 Bleaching Event at Lee Stocking Island, Bahamas. Coral Reefs 28 (1): 209–14. https://doi.org/10.1007/s00338-008-0455-7.
Cunning, Ross, Rachel N. Silverstein, and Andrew C. Baker. 2018. Symbiont Shuffling Linked to Differential Photochemical Dynamics of Symbiodinium in Three Caribbean Reef Corals. Coral Reefs 37 (1): 145–52. https://doi.org/10.1007/s00338-017-1640-3.
Finelli, C. M., B. S. Helmuth, N. D. Pentcheff, and D. S. Wethey. 2007. Intracolony Variability in Photosynthesis by Corals Is Affected by Water Flow: Role of Oxygen Flux. Marine Ecology Progress Series 349: 103–10. https://doi.org/10.3354/meps07101.